In 1997, when Deep Blue defeated chess champion Garry Kasparov, artificial intelligence was still confined to narrow, specialized tasks. Today, AI writes poetry, diagnoses diseases, and helps design rocket engines. This isn’t just another technology—it’s the first time we’ve automated intelligence itself, at scale.
Yet for investors, AI presents a paradox. It’s undeniably impressive, but translating technical breakthroughs into sustainable business value remains a challenge. Some AI companies could become the next Apple or Google. Others will undoubtedly overpromise and collapse. This primer cuts through both the skepticism and the hype to examine where real economic value emerges. We’ll explore:
✔ The exponential mechanisms driving AI growth
✔ How the AI technology stack creates—and captures—value
✔ Which phases of AI adoption create investable opportunities
✔ Where the greatest risks and rewards concentrate
The difference between AI winners and losers won’t just be technical excellence. It will be their ability to convert algorithmic advances into defensible business models. Let’s examine how.
The Exponential Mechanism
Artificial intelligence follows an exponential trajectory not because it’s “the future” or because it’s vaguely “disruptive,” but due to a set of tightly interlocking feedback loops. Let’s review each one.
Compounding Intelligence
In traditional software, a developer writes code, and the program remains static. AI, by contrast, reinvests every new interaction: more data leads to higher accuracy, which leads to greater adoption, which generates even more data.
For example, before AI, Google’s search engine relied on hand-coded rules to rank web pages. It used signals like keyword frequency, backlinks, and page structure. While effective, this system was static—if a new type of query emerged, engineers had to manually tweak the algorithm.
In 2015, Google introduced RankBrain, a machine learning system that improves over time based on user interactions. Here’s how it works:
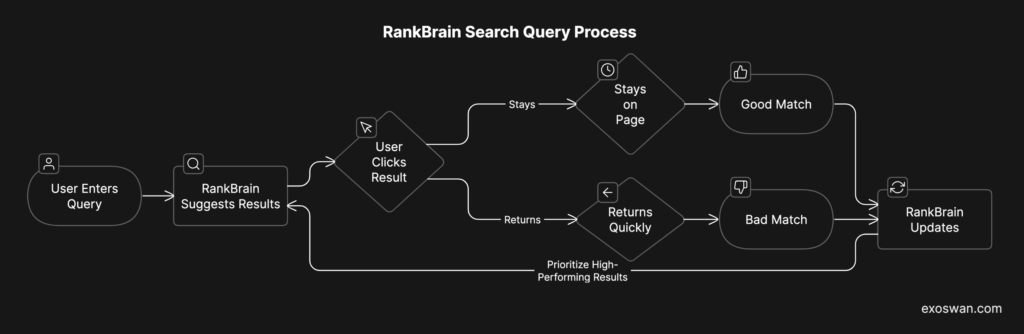
- A user enters a new, complex search query (e.g., “best laptop for video editing under $1,500”).
- RankBrain suggests results based on similar queries, even if no exact match exists.
- The user’s behavior feeds back into the system:
- If they click a result and stay on the page, RankBrain interprets it as a good match.
- If they click and quickly return to search, RankBrain assumes it was a bad match.
- RankBrain updates itself dynamically—prioritizing high-performing results for future searches.
Each new query teaches RankBrain to improve its predictions, with no manual adjustments needed. This is a self-reinforcing economic engine. More users make the system smarter, and a smarter system attracts more users. This compounding intelligence isn’t unique to Google—it underpins all AI-driven business models:
- Tesla’s Autopilot: More cars on the road → More driving data → Better AI-assisted driving → Safer performance → More adoption → More data.
- TikTok’s Algorithm: More engagement → More personalized content recommendations → Higher user retention → More content consumption → Smarter AI.
- ChatGPT & AI Assistants: More interactions → More fine-tuned responses → More user trust → More usage → More improvement.
As AI companies continue to operate, they accumulate a data and learnings moat that’s incredibly difficult for new entrants to compete with. That’s why AI is often described as a “race”—it’s a winner-takes-most market.
The Cost Curve of Intelligence
AI is the first technology to automate thinking itself. Just as machines reduced the cost of physical labor, AI slashes the cost of cognitive work—analyzing data, writing reports, coding software. And like all digital technologies, once an AI model is trained, its marginal cost approaches zero.
For example, generative AI models can produce thousands of legal contracts or customer service responses in seconds. Yet the underlying model was trained only once. This is a sharp departure from traditional knowledge work, where more output requires more labor.
The paradigm shift is that intelligence—previously expensive and scarce—is being converted into something cheap and abundant. For many industries, the cost of not adopting AI is now higher than the cost of implementation.
The Hardware–Software Flywheel
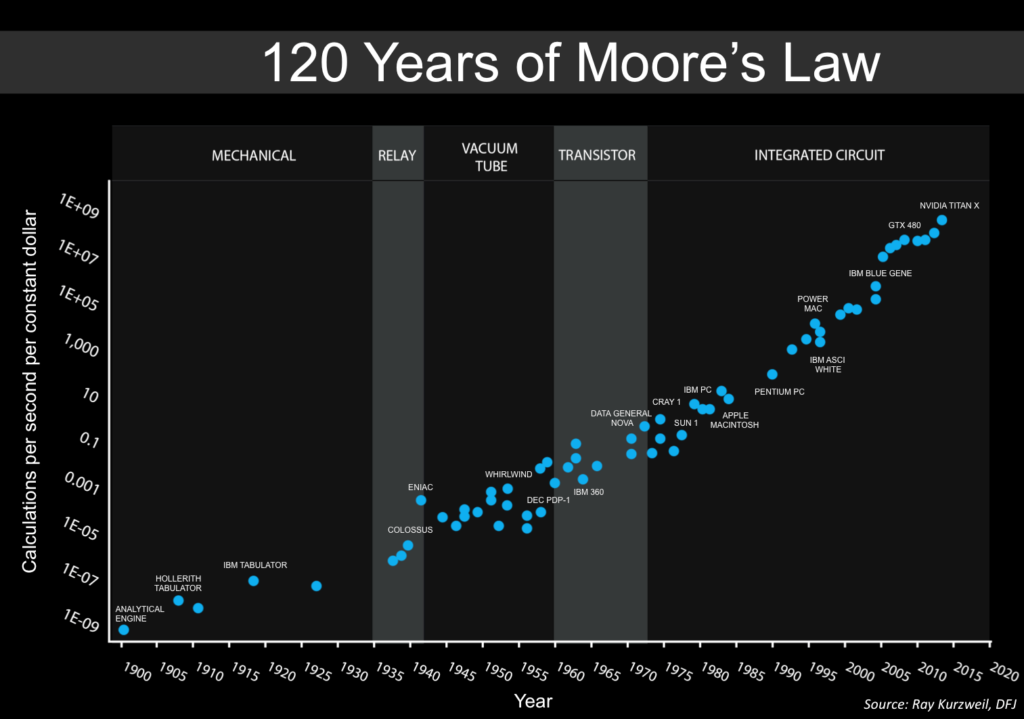
AI advances are not driven by software alone. They are intertwined with exponential improvements in computing hardware. Moore’s Law (doubling of transistors roughly every two years) and the rise of AI-specific chips (GPUs, TPUs, and custom silicon) mean that the cost of running large AI models keeps dropping. This creates a flywheel: better chips enable bigger models, bigger models demand better chips.
The Automation of AI Development Itself
Perhaps the most profound driver of AI’s exponential growth is that AI is now helping to build better AI. Techniques like reinforcement learning, neural architecture search, and self-supervised learning allow AI models to optimize their own parameters, design new algorithms, and even code improved versions of themselves.
Take reinforcement learning for example:
Imagine you have an AI that needs to learn to play the Atari game Breakout (where a paddle bounces a ball to break bricks). The AI starts with no understanding of the game—just raw pixel inputs from the screen and the ability to move the paddle left or right.

Reinforcement learning is structured around trial and error, where the AI agent learns by interacting with the environment and receiving rewards based on its actions.
- Observation – The AI sees the game screen as raw pixel data.
- Action – The AI moves the paddle left or right.
- Reward – If the ball hits a brick, the AI gets a positive reward (e.g., +1 point). If the ball falls past the paddle, the AI gets a negative reward (e.g., -1 point).
- Learning from Rewards – Over time, the AI adjusts its actions to maximize the total score.
The core idea is that good actions get reinforced, while bad actions get suppressed, just like how a human might learn through experience.
At the start, the AI plays poorly—missing the ball frequently. But as it plays thousands of games, it learns to position the paddle just right, controlling the ball’s trajectory.
Eventually, though, it finds a “superhuman strategy.” It aims the ball to carve tunnels through bricks, trapping it at the top of the screen for continuous hits—something many human players never discover.
That’s because AI isn’t just learning better—it learns differently. Human players often focus on short-term ball control. AI, through sheer iteration volume, finds non-obvious strategies that maximize score over the long term.
The State of AI Technology
Artificial intelligence isn’t a magic bullet. And contrary to what the some people in the media would have you believe, we’re nowhere close to AI “taking over everything.” For investors, it’s critical to distinguish between the future promises and the current realities of AI.
AI That Works Today
The key question right now is not, “where is AI being used?” Instead, it’s “where does AI create durable and valuable competitive advantages?” This distinction is the difference between adoption for the sake of it (i.e. hopping on the bandwagon) and actual value creation. Let’s look at a few key areas of the latter:
Generative AI in Creative Work & Coding
Generative AI is rapidly reshaping digital productivity. Large language models (LLMs), image generators, and AI coding assistants are becoming integral parts of professional workflows. Take software engineering, for example. Traditionally, developers spent a significant portion of their time writing boilerplate code, debugging, and refactoring. AI-assisted coding tools like GitHub Copilot and Replit Ghostwriter are dramatically reducing this workload. Some companies have seen output gains of almost 40% since adopting AI-assisted coding tools.
This transformation is changing the economics of software development. Large enterprises can deploy AI copilots across teams, but the more disruptive impact is in the startup world. For example, a small fintech startup that would have previously needed a 10-person engineering team to build a full-stack banking platform can now achieve similar results with just three engineers using AI-assisted development. This lowers the barrier to entry, enabling early-stage startups to compete at lower costs.
Search and Recommendation Systems
Search and recommendation engines are not just AI-powered features—they are core economic drivers for search engines, e-commerce platforms, and social media feeds. For example, AI-driven search in Google, Amazon, and e-commerce marketplaces optimizes results based on behavioral signals—click-through rates, time spent on page, past purchases—ensuring results are ranked not just by relevance but by likelihood to convert into a purchase or engagement.
Meanwhile, AI recommendation systems drive engagement on platforms like TikTok, Netflix, and YouTube by analyzing how users interact with content. TikTok’s For You page is a prime example: its AI tracks how long users watch each video, whether they replay it, swipe away instantly, or engage with comments. If a user watches multiple cooking videos to completion, the algorithm rapidly fills their feed with more recipe content—even tailoring recommendations down to specific cuisines. Similarly, Netflix fine-tunes its recommendations not just based on viewing history but on detailed metrics like pause frequency, rewind behavior, and even the types of thumbnails users are more likely to click.
Codifying Human Judgement
AI excels at high-volume, pattern-based decision-making, especially in domains where historical data can predict future outcomes with high accuracy. This is why it has revolutionized fraud detection in financial services. Payment networks like Visa and Mastercard process over 500 million transactions per day, and even a 0.01% reduction in fraudulent transactions translates to hundreds of millions of dollars in savings annually.
Another critical area where AI codifies human judgment is medical diagnostics. Radiologists interpret thousands of medical images each year, looking for subtle signs of diseases like cancer, fractures, or neurological conditions. AI models trained on millions of X-rays, MRIs, and CT scans can now detect abnormalities with accuracy comparable to—or in some cases, exceeding—that of human specialists. Google’s DeepMind AI, for instance, has demonstrated superior accuracy in diagnosing eye diseases from retinal scans.
AI That’s On Its Way
Some AI capabilities are not fully solved yet but are advancing quickly. Investors should watch these closely, as the companies leading them could unlock massive value.
One of the most promising frontiers is AI-driven autonomous task execution. Today’s AI assistants are impressive but still require handholding and human oversight. The next step is AI agents that can operate independently, handling multi-step tasks without constant user input. Instead of just answering emails, future AI systems could scan an inbox, categorize messages by priority, draft responses, schedule meetings, and even coordinate cross-team projects—essentially functioning as a virtual chief of staff.
Another rapidly advancing area is AI’s ability to reason, plan, and make strategic decisions. Today’s models excel at pattern recognition and summarization but struggle with causality, long-term planning, and decision-making under uncertainty. These are key skills needed for AI to be effective in law, medicine, and business strategy. Researchers are exploring hierarchical AI systems, reinforcement learning with memory, and multi-agent collaboration, which could enable AI to handle complex legal arguments, long-term financial portfolio management, or even diagnose rare medical conditions with greater accuracy.
Self-driving cars remain a high-profile but partially solved problem. AI can navigate highways and controlled urban environments with high accuracy, but edge cases—such as unexpected roadwork, erratic pedestrians, or extreme weather—still pose challenges. Companies like Waymo, Cruise, and Tesla have deployed autonomous taxis in limited settings, such as San Francisco, Phoenix, and Las Vegas, but scaling to full Level 5 autonomy (no human intervention required) remains elusive. The biggest barriers are not just technical but regulatory and liability concerns, as governments struggle to set safety standards for self-driving fleets.
AI That Remains Unsolved
Some AI challenges remain fundamentally unsolved and may take years—if not decades—to crack. Investors should be cautious of companies making overhyped claims in these areas.
The most speculative frontier is Artificial General Intelligence (AGI)—the idea of AI that can think, reason, and act across any domain like a human. Despite rapid advancements, today’s AI remains a sophisticated pattern-matching system, not a true reasoning entity. While researchers are experimenting with cognitive architectures like hybrid neural-symbolic reasoning and memory-augmented models, there is no clear roadmap or timeline for when—or if—AGI will be achieved. Some experts believe it’s a matter of decades, while others argue it may never happen.
Another major unsolved challenge is AI’s massive energy consumption. Cutting-edge AI models require enormous computational power, leading to skyrocketing energy demands. Training GPT-4, for example, likely consumed millions of kilowatt-hours—comparable to the yearly energy usage of a small town. As AI adoption accelerates, data centers are experiencing a bottleneck in power availability, forcing companies to invest in nuclear power and alternative energy sources to sustain their AI operations. Without breakthroughs in hardware efficiency, AI’s energy footprint could become an economic and environmental constraint.
A related issue is the physical limits of AI scaling. The success of large AI models has been largely driven by brute-force scaling—bigger datasets, more GPUs, and larger neural networks. However, this approach faces diminishing returns. Chip shortages, supply chain constraints, and the sheer cost of running multi-billion parameter models make continued scaling unsustainable. Researchers are exploring post-Moore’s Law computing paradigms, such as neuromorphic chips, optical computing, and quantum AI, but these technologies are still in their infancy. Without a paradigm shift in computing, the current AI boom could plateau as hardware limitations catch up.
Market Landscape: The AI Tech Stack
Artificial intelligence isn’t a monolithic technology. Instead, it’s best thought of as a stack of interlocking layers that come together to form the broader AI market:
Compute Layer
At the bottom of the stack is compute power—the raw hardware that makes AI possible. AI models require enormous computational resources, both for training (building models) and inference (running them in real-world applications).
- Training AI models is highly compute-intensive, often requiring specialized processors designed for parallel workloads. These models are trained on vast datasets, sometimes over weeks or months.
- Inference—the process of applying a trained model to real-world tasks—is less intensive but must happen at scale and in real-time, especially for applications like chatbots, recommendation systems, or autonomous vehicles.
Challenges: Compute costs are a major bottleneck. Training large AI models can cost millions of dollars in energy and hardware expenses, and inference costs scale exponentially with usage. As demand for AI grows, access to high-performance computing becomes a competitive constraint.
Examples:
| Company | Focus | Key Differentiators |
|---|---|---|
| NVIDIA | AI GPUs & Accelerators | Dominates AI chips with CUDA software and high-performance GPUs. |
| AMD | CPUs & GPUs for AI | Competes with NVIDIA, offering AI-optimized processors and cost-effective alternatives. |
| Cerebras Systems | AI-Specific Processors | Builds wafer-scale chips for extreme AI training efficiency. |
| Custom AI Chips (TPUs) | Optimizes TPUs for AI workloads, powering Google’s cloud and AI services. | |
| AWS | Cloud AI Compute | Provides AI-optimized cloud infrastructure with custom Trainium chips. |
Data Layer
AI models are only as good as the data they’re trained on. The data layer includes everything from raw datasets to data preprocessing and storage systems.
- Structured data (financial transactions, medical records, sensor logs) is easier to process but often limited in scope.
- Unstructured data (text, images, video, audio) is far more abundant but requires heavy preprocessing and annotation to be useful for AI.
- Real-time data is critical for AI applications like fraud detection, autonomous systems, and personalized recommendations.
Challenges: High-quality, domain-specific data is scarce and expensive to acquire. Companies must balance data privacy, regulatory compliance, and bias mitigation—especially in industries like healthcare and finance. Ownership and control over proprietary data can create strong competitive moats, but also regulatory risk.
Examples:
| Company | Focus | Key Differentiators |
|---|---|---|
| Databricks | Unified Data Analytics Platform | Integrates data engineering, analysis, and AI workloads into a single platform, streamlining workflows. |
| Qlik | Data Integration and Analytics | Offers associative data indexing, enabling users to explore data relationships across multiple sources intuitively. |
| Palantir Technologies | Data Management and Security | Provides platforms that integrate, manage, and secure vast datasets, facilitating complex data analysis. |
| Dataiku | Enterprise AI and Machine Learning | Delivers a collaborative environment for data professionals to develop, deploy, and monitor AI models efficiently. |
| Alteryx | Data Preparation and Analytics Automation | Simplifies data preparation and blending processes, enabling automated analytics workflows for faster insights. |
Models & Algorithms Layer
The models themselves sit at the core of the AI stack. These are the neural networks, transformers, and statistical learning models that process inputs and generate intelligent outputs.
- Foundation Models (large, general-purpose models) are trained on massive datasets and adapted for multiple tasks—such as text generation, code writing, or image recognition.
- Domain-Specific Models are fine-tuned for specialized applications—such as fraud detection in banking or medical imaging in healthcare.
- Optimization Algorithms improve performance by making models more efficient, reducing computation costs, and enabling deployment on lower-power devices.
Challenges: The biggest challenge here is scalability vs. efficiency—larger models deliver better performance but are costly to train and run. Model explainability and bias also remain unresolved problems, limiting AI adoption in highly regulated sectors.
Examples:
| Company | Focus | Key Differentiators |
|---|---|---|
| OpenAI | Large Language Models | Pioneers in developing advanced language models, enabling natural language understanding and generation. |
| Google DeepMind | Reinforcement Learning | Specializes in reinforcement learning, achieving breakthroughs in complex problem-solving and game-playing AI. |
| Anthropic | AI Safety and Alignment | Focuses on creating AI systems that are interpretable and aligned with human values, emphasizing safety in AI development. |
| Cohere | Natural Language Processing | Develops language models tailored for enterprise applications, enhancing business-specific language understanding. |
| Hugging Face | Open-Source AI Models | Provides a platform for sharing and collaborating on open-source AI models, fostering community-driven AI development. |
Deployment Layer
This layer includes platforms and services that help companies deploy AI models at scale—whether through cloud-based AI services, enterprise AI platforms, or low-code AI tools. These companies bridge the gap between AI research and real-world implementation by providing APIs, AI model hosting, and workflow automation.
- Cloud-Based AI Services provide scalable computing power, enabling businesses to train and deploy AI models without owning infrastructure.
- Enterprise AI Integration Platforms allow organizations to embed AI into their existing workflows, offering pre-trained models and automation tools.
- AI Development Frameworks simplify the process of building and fine-tuning AI models, often including low-code/no-code options for non-technical users.
Challenges: AI deployment remains compute-intensive, making cloud services expensive at scale. Security and data privacy concerns are critical, particularly in regulated industries like healthcare and finance. Additionally, many AI models work well in research but struggle in real-world environments, requiring extensive fine-tuning and ongoing maintenance.
Examples:
| Company | Focus | Key Differentiators |
|---|---|---|
| Amazon Web Services (AWS) | Cloud-Based AI Services | Provides AI/ML tools for scalable deployment, including custom AI accelerators. |
| Microsoft Azure AI | Enterprise AI Integration | Enables seamless AI adoption with pre-built models and deep enterprise software integration. |
| Google Cloud AI | End-to-End AI Solutions | Offers a full AI ecosystem, including AutoML, data pipelines, and AI-driven analytics. |
| IBM Watsonx | AI for Business Applications | Focuses on enterprise AI solutions with strong regulatory compliance and explainability features. |
| C3 AI | Enterprise AI Application Development | Provides a development platform for AI-driven enterprise applications across industries. |
Application Layer
This layer consists of AI-first applications that leverage AI to solve industry-specific problems, spanning sectors like healthcare, finance, media, and automation.
- AI Agents & Workflow Automation streamline business operations by automating complex, multi-step tasks across software tools.
- Generative AI for Media & Content enables AI-driven video editing, image creation, and automated storytelling, transforming creative workflows.
- AI in Healthcare & Life Sciences applies machine learning to medical imaging, drug discovery, and personalized treatment recommendations.
- AI-Driven Fintech & Lending improves credit risk modeling, fraud detection, and automated decision-making for financial institutions.
Challenges: AI applications in regulated industries face legal and ethical risks, from biased lending models to AI-driven misdiagnoses. Scalability is also a challenge, as many AI startups struggle to transition from prototype to commercial-scale deployment. Finally, reliability and explainability remain barriers to adoption, particularly in fields where trust and accountability are critical.
Examples:
| Company | Focus | Key Differentiators |
|---|---|---|
| Adept AI | AI Agents & Workflow Automation | Builds AI agents that can take actions across software tools, automating complex workflows. |
| Runway ML | Generative AI for Video | Specializes in AI-driven video editing and content creation tools for media professionals. |
| Hugging Face | AI Model Collaboration | Provides an open-source platform for sharing and fine-tuning AI models across industries. |
| Upstart | AI-Powered Lending | Uses AI to assess creditworthiness beyond traditional FICO scores, improving loan approvals. |
| Tempus | AI in Healthcare & Genomics | Applies AI to medical data for personalized treatment plans and drug discovery. |
Investment Implications
When evaluating AI companies, it’s important to distinguish them from traditional software businesses. The mistake many investors make is focusing on raw model performance or technical innovation alone. However, their long-term value depends not just on technical breakthroughs, but also on their ability to defend against commoditization, scale efficiently, and embed AI into critical workflows.
Remember: AI moves fast, and today’s best models won’t necessarily win in the long run. Instead, consider these three core factors to determine whether an AI company has staying power:
Proprietary Data Loops
Who owns the learning flywheel? As discussed earlier, AI’s compounding value comes from self-reinforcing data advantages. The strongest companies:
- Generate proprietary data as a byproduct of usage (e.g., Tesla’s fleet, TikTok’s engagement patterns)
- Improve automatically with scale and feedback loops
- Face no easy replication by competitors, even with similar models
🚩 Red Flag: AI companies relying entirely on third-party models or publicly available datasets—they lack an enduring moat.
Unit Economics at Scale
Who can monetize without margin compression? Many AI companies struggle to turn technological breakthroughs into sustainable profits. Strong AI companies:
- Have scalable revenue models (e.g., API-based pricing, enterprise subscriptions)
- Manage compute costs effectively, either through custom infrastructure or model optimizations
- Are able to pass higher costs onto customers due to high switching costs or must-have functionality
🚩 Red Flag: AI companies where margins shrink as they scale due to rising inference costs or reliance on expensive API calls.
Workflow Integration
Who becomes a systemic part of business operations? AI-driven automation only creates lasting value if it becomes deeply embedded in workflows, making it costly or impractical to remove. Strong AI companies:
- Integrate directly into existing business processes (e.g., AI copilots for software development, AI-driven underwriting in finance)
- Have high switching costs—customers don’t just use the tool; they rely on it
- Show signs of expanding use cases within existing customer accounts
🚩 Red Flag: AI companies selling tools that are easily replaceable or do not become a mission-critical part of operations.
Putting It All Together
Artificial intelligence is an evolving market that will mature in phases, each with different risks and opportunities. Keep in mind that AI investing isn’t about picking the best models. Don’t just bet on AI—bet on moats:
🔹 Proprietary Data Loops → Defensible self-improving models
🔹 Unit Economics at Scale → AI that doesn’t erode margins
🔹 Workflow Integration → Deep, sticky adoption
This is an evolving market that will mature in phases, each with different risks and opportunities:
Phase 1: Infrastructure & Model Arms Race (Now – 3 Years)
Right now, AI investment is dominated by infrastructure players and foundational model developers. This phase is about building the plumbing of AI: compute, data pipelines, and foundational models.
Key Investment Themes:
- Compute Scarcity Creates Pricing Power → AI workloads require immense compute resources, benefiting chipmakers, cloud providers, and specialized hardware developers. The real winners are those who control AI compute access.
- Foundation Models are Capital-Intensive, with Uncertain Moats → Leading AI labs are burning billions to train larger and more powerful models, but monetization remains a challenge. Early leaders may not maintain dominance if smaller, more efficient models disrupt the space (e.g., DeepSeek).
- Middleware & AI Dev Tools are the Quiet Winners → AI application development is bottlenecked by complexity—whoever simplifies AI deployment will capture sticky, recurring revenue.
Phase 2: AI-Driven Enterprise Transformation (3-7 Years)
As AI infrastructure stabilizes, the focus will shift to business adoption and industry-specific AI applications. This phase will see AI embedded into enterprise workflows and transforming decision-making across sectors.
Key Investment Themes:
- AI-Native Business Models Will Emerge → The most valuable companies won’t just be those using AI—they’ll be built around AI automation, decision intelligence, and adaptive learning systems.
- Regulatory & Compliance AI Will Be a Must-Have → AI’s expansion into finance, healthcare, and law will require transparent, explainable, and compliant AI solutions. Expect growth in AI safety, auditing, and governance tools.
- Vertical AI Will Outperform General AI → Industry-specific AI models fine-tuned for legal, medical, and financial domains will outperform general-purpose models in terms of accuracy and trustworthiness.
Phase 3: AI as an Autonomous Economic Agent (7+ Years)
The final stage is when AI transitions from being a tool for augmentation to an autonomous driver of economic activity. This is where AI starts making high-level decisions, operating businesses, and acting independently in markets.
Key Investment Themes:
- AI-Run Companies Will Exist → Entirely AI-driven businesses—with minimal human oversight—will emerge, leveraging AI agents to manage operations, trading, and even entire supply chains.
- Synthetic Data & Simulated Economies → AI will start generating its own training data and testing environments, making real-world data dependencies less critical. This will unlock entirely new AI-driven industries.
- Post-Labor Economics & New Market Structures → If AI eliminates the need for human-driven decision-making in key industries, it will reshape corporate structures, workforce dynamics, and capital allocation strategies.
Investment Strategy
To navigate AI’s evolution, match your approach to its maturity curve. Consider a barbell strategy:
- Core Positions: AI infrastructure leaders (compute, cloud, and foundational models) with high pricing power today.
- Growth Exposure: AI-native companies building sticky enterprise AI applications and automation solutions.
- Moonshots: AI-first companies experimenting with autonomy, AI-driven businesses, and synthetic data ecosystems.
The biggest mistake investors make is treating AI as a single wave of disruption. AI isn’t just another tech cycle—it’s a paradigm shift that will unfold in stages, each with distinct risks and rewards.
Remember, the real question isn’t just who is building AI—it’s who is building around AI in ways that create enduring, compounding advantages. Those are the investments that will define the next decade.